Research Article
Quantitative Trait Loci Analysis in Triticeae: Implications for Breeding and Genetics 
 Author
Author  Correspondence author
Correspondence author
Triticeae Genomics and Genetics, 2024, Vol. 15, No. 4 doi: 10.5376/tgg.2024.15.0018
Received: 28 May, 2024 Accepted: 05 Jul., 2024 Published: 18 Jul., 2024
Zhang G.P., and Cai R.X., 2024, Quantitative trait loci analysis in Triticeae: implications for breeding and genetics, Triticeae Genomics and Genetics, 15(4): 185-195 (doi: 10.5376/tgg.2024.15.0018)
Quantitative Trait Loci (QTL) analysis has become a pivotal tool in understanding the genetic basis of complex traits in Triticeae, with significant implications for breeding and genetics. This study retrospects various methodologies and findings in QTL mapping, highlighting the advancements and applications in crop improvement. Multiple interval mapping (MIM) has been shown to enhance the precision and power of QTL detection, allowing for the estimation of epistasis, genotypic values, and heritabilities. Comparative genomic analysis has identified stable QTLs for grain yield, quality traits, and micronutrient contents in wheat, consolidating QTLs into meta-QTLs (MQTLs) and reducing confidence intervals, thus facilitating marker-assisted selection (MAS). The integration of linkage and association mapping has further dissected the genetic architecture of complex traits, revealing the importance of environment-specific QTLs and the need for advanced statistical strategies in the era of next-generation sequencing. Additionally, QTL mapping under stress conditions, such as salinity, has identified key genomic regions contributing to stress tolerance, which can be exploited for breeding resilient varieties. ThFe development of multiparental populations and the use of whole-genome resequencing (QTL-seq) have accelerated the identification of QTLs, providing a robust framework for genetic dissection and crop improvement. This study underscores the critical role of QTL analysis in advancing our understanding of genetic variation and enhancing breeding programs in Triticeae.
1 Introduction
The Triticeae tribe, which includes economically significant cereals such as wheat, barley, and rye, is fundamental to global agriculture. These crops are cultivated extensively for their grain, which serves as a staple food source for a large portion of the world's population. The genetic diversity within Triticeae is vast, encompassing numerous species and varieties that have adapted to a wide range of environmental conditions. This diversity is a valuable resource for breeding programs aimed at improving crop yield, disease resistance, and stress tolerance.
Quantitative Trait Loci (QTL) are regions of the genome that are associated with the variation of quantitative traits, which are typically influenced by multiple genes and environmental factors. QTL mapping is a powerful tool used to identify these genomic regions and understand the genetic architecture of complex traits. Traditional QTL mapping involves the use of biparental populations, but recent advancements have seen the development of multiparental cross designs, which offer broader genetic bases and higher mapping resolution (Crepieux et al., 2004; Milner et al., 2016). Techniques such as Bayesian mapping and composite interval mapping have further enhanced the precision and power of QTL detection (Jiang and Zeng, 1995; Yi and Xu, 2001; Takagi et al., 2013). These methodologies are crucial for dissecting the genetic basis of traits such as yield, stress tolerance, and nutrient content in crops (Zhang et al., 2010; Ilyas et al., 2019; Shariatipour et al., 2021).
The study conducts a comprehensive QTL analysis in Triticeae, with a focus on identifying loci associated with key agronomic traits. By leveraging advanced QTL mapping techniques and multiparental populations, this research aims to uncover the genetic factors that contribute to trait variation and their interactions with environmental conditions. The findings will have significant implications for breeding programs, providing valuable insights for the development of improved Triticeae varieties with enhanced performance and resilience. This study also seeks to explore the potential of marker-assisted selection (MAS) in accelerating the breeding process by utilizing identified QTLs for targeted trait improvement.
2 Methodological Advances in QTL Analysis
2.1 Traditional QTL mapping techniques
Traditional QTL mapping techniques have laid the foundation for understanding the genetic basis of quantitative traits. These methods typically involve the use of biparental populations, where two parent lines are crossed to produce a mapping population. The phenotypic data of the progeny, along with their genotypic data, are analyzed to detect associations between markers and traits. Early methods, such as single-marker analysis and interval mapping, were based on simple statistical models like analysis of variance (ANOVA) and regression analysis (Doerge, 2002; Singh and Singh, 2015).
Single QTL mapping methods detect one QTL at a time, which can be limiting when dealing with complex traits controlled by multiple loci. To address this, multiple QTL mapping (MQM) was developed, combining multiple regression analysis with interval mapping to include all significant QTLs in the genetic model (Singh and Singh, 2005). Composite interval mapping (CIM) further improved the precision of QTL detection by incorporating background genetic variation into the model, thus increasing the power to detect QTLs and estimate their effects more accurately (Jiang and Zeng, 1995; Singh and Singh, 2015).
2.2 Modern QTL mapping approaches
Modern QTL mapping approaches have evolved to address the limitations of traditional methods, particularly in terms of resolution and the ability to handle complex traits. One significant advancement is the use of multiparental populations, such as recombinant inbred lines (RILs) derived from multiple parents. These populations provide a broader genetic base and higher mapping resolution compared to biparental populations. For instance, a study on durum wheat utilized a RIL population derived from four elite cultivars, resulting in the identification of 16 QTLs across different environments and detection methods (Figure 1) (Milner et al., 2016).
 Figure 1 Figure 1 Proportion of 338 RILs assigned to each of the four founders across the NCCR linkage map (Adapted from Milner et al., 2016) Image caption: The chart shows that the probabilities of the founders vary across different chromosome regions, indicating that each founder contributes differently to different chromosomes. For example, Claudio and RasconTarro show significant probability fluctuations across multiple chromosomes, while Colosseo and Neodur have relatively stable probabilities. Overall, the probabilities for all founders range between 0.15 and 0.35. These probability curves help to understand each founder's genetic contribution across the genome, which is significant for breeding and genetic analysis (Adapted from Milner et al., 2016) |
Another modern approach is the use of joint analysis of multiple traits, which takes into account the correlated structure of traits to improve the statistical power and precision of QTL detection. This method allows for the testing of biologically interesting hypotheses, such as pleiotropy versus close linkage, and can provide insights into the genetic correlations between different traits (Jiang and Zeng, 1995).
Meta-QTL analysis is another powerful tool that integrates results from multiple QTL studies to identify stable and reliable QTLs. This approach reduces the confidence intervals of QTLs, making it easier to pinpoint candidate genes. For example, a meta-analysis in wheat condensed 735 QTLs into 100 meta-QTLs, significantly narrowing down the genomic regions of interest (Shariatipour et al., 2021).
2.3 Technological innovations in QTL analysis
Technological innovations have revolutionized QTL analysis, making it faster, more accurate, and more comprehensive. One such innovation is QTL-seq, which combines bulked segregant analysis with whole-genome resequencing. This method involves sequencing the DNA of two groups of individuals with extreme phenotypes and identifying QTLs based on the differences in allele frequencies between the groups. QTL-seq has been successfully applied in rice to rapidly identify QTLs for traits like disease resistance and seedling vigor (Takagi et al., 2013).
Another significant technological advancement is the use of high-density genetic maps and next-generation sequencing (NGS) technologies. These tools provide high-resolution mapping and enable the identification of QTLs at a subcentimorgan scale. For instance, fine mapping using selected overlapping recombinant chromosomes has been employed in tomato to map QTLs to very small intervals, facilitating the identification of candidate genes (Paterson et al., 1990).
The development of software tools and statistical models has also played a crucial role in advancing QTL analysis. Software packages like R/qtl allow researchers to perform multiple QTL mapping and other complex analyses with ease (Powder, 2020). Additionally, new methods for QTL validation, such as the use of near-isogenic lines (NILs) and intercross recombinant inbred lines, have improved the accuracy and reliability of QTL studies (Singh and Singh, 2015).
The field of QTL analysis has seen significant methodological advances, from traditional mapping techniques to modern approaches and technological innovations. These advancements have enhanced our ability to dissect complex traits, identify candidate genes, and apply this knowledge to breeding programs for crop improvement. The integration of high-resolution mapping, multiparental populations, and advanced statistical models continues to push the boundaries of what is possible in QTL analysis, offering new opportunities for genetic research and practical applications in agriculture.
3 QTLs in Triticeae: Case Studies and Applications
3.1 QTL mapping in wheat (Triticum aestivum)
Quantitative Trait Loci (QTL) mapping in wheat has been instrumental in identifying genomic regions associated with key agronomic traits, which are crucial for improving yield and stress tolerance. For instance, a comprehensive global QTL analysis identified stable loci for yield-related traits on chromosomes 4A and 4B in the Nongda3338/Jingdong6 doubled haploid population. This study revealed significant trade-offs between thousand grain weight (TGW) and grain number per spike (GNS) on chromosome 4A, and identified a novel QTL for heat susceptibility index of TGW on chromosome 4BL, which explains approximately 10% of phenotypic variation (Börner et al., 2002).
Another study conducted a meta-analysis of 735 QTLs from 27 independent mapping populations, consolidating them into 100 meta-QTLs (MQTLs) distributed across wheat chromosomes. This analysis highlighted the co-localization of QTLs for grain yield (GY) and grain protein content (GPC), with significant overlaps found on chromosomes 2A, 3B, and 4D. The study also identified candidate genes within these MQTLs, providing insights into the genetic mechanisms driving quantitative variation for these traits (Figure 2) (Shariatipour et al., 2021).
 Figure 2 Distribution of projected quantitative trait loci (QTLs) across the wheat (Triticum aestivum L.) chromosomes (Adopted from Shariatipour et al., 2021) Image caption: The numbers inside each parenthesis represent the number of QTLs (Adopted from Shariatipour et al., 2021) |
In the spring wheat cross 'Louise'×'Penawawa', QTLs associated with seedling growth habit, leaf color, plant height, flowering date, maturity date, grain volume weight, grain protein content, and grain yield were mapped. Notably, QTLs for flowering date and maturity date were linked to the PpdD1 gene for photoperiod insensitivity, while QTLs for plant height were localized to chromosomes 2D and 3B, demonstrating the pleiotropic effects of the PpdD1 gene (Guan et al., 2018).
3.2 QTL analysis in barley (Hordeum vulgare)
Barley, another important member of the Triticeae tribe, has also been the focus of extensive QTL mapping studies. These studies have aimed to improve traits such as yield, disease resistance, and stress tolerance. For example, a high-density genetic map of barley was used to identify QTLs for yield and yield components under various environmental conditions. This map revealed clusters of QTLs on chromosomes 1H, 2H, and 3H, which were associated with traits like grain number per spike and thousand grain weight (Backes et al., 1995).
In another study, QTLs for agronomic traits such as plant height, flowering time, and grain yield were mapped in a barley population derived from a cross between two elite cultivars. The study identified several QTLs with significant effects on these traits, providing valuable markers for marker-assisted selection (MAS) in barley breeding programs (Tshikunde et al., 2019).
3.3 QTL studies in other Triticeae species
Beyond wheat and barley, QTL mapping has been applied to other Triticeae species to enhance our understanding of their genetic architecture and improve their agronomic performance. For instance, in intermediate wheatgrass (Thinopyrum intermedium), a perennial grass related to wheat, QTL mapping identified 111 QTLs associated with traits such as seed size, shattering, threshing, and fertility. This study highlighted the potential of using QTLs to accelerate the domestication of intermediate wheatgrass for dual-purpose forage and grain production (Ilyas et al., 2019).
In hexaploid wheat, a study involving a population of 96 doubled haploid lines from the cross Chinese Spring × SQ1 identified QTLs for yield and yield components across various stress conditions. The study found that yield QTLs were widely distributed around the genome, with significant clusters on chromosomes 1D, 4B, and 7A. These findings underscore the importance of QTL mapping in identifying genomic regions that contribute to yield stability under different environmental stresses (Backes et al., 1995).
Additionally, advanced backcross QTL (AB-QTL) analysis has been used to transfer favorable alleles from wild relatives of wheat into elite varieties. For example, a study involving a BC2F2 population derived from a cross between the German winter wheat variety 'Prinz' and a synthetic wheat line identified 40 putative QTLs for yield and yield components. This approach demonstrated the potential of using wild relatives to enhance the genetic diversity and agronomic performance of cultivated wheat (Tshikunde et al., 2019).
Overall, QTL mapping in Triticeae species has provided valuable insights into the genetic basis of important agronomic traits, facilitating the development of improved varieties through marker-assisted selection and other breeding strategies. The integration of QTL mapping with advanced genomic tools continues to enhance our ability to dissect complex traits and apply this knowledge to crop improvement programs.
4 Implications for Triticeae Breeding and Genetics
4.1 Marker-assisted selection (MAS)
Marker-Assisted Selection (MAS) has revolutionized the field of plant breeding by significantly enhancing the precision and efficiency of selecting desirable traits. In Triticeae, which includes important cereal crops like wheat and barley, MAS has been instrumental in accelerating the breeding process and improving the accuracy of trait selection. The integration of DNA markers with traditional breeding methods allows for the identification and selection of plants carrying beneficial alleles at an early stage, thus reducing the time and resources required to develop new varieties (Mohan et al., 1997; Hasan et al., 2021).
MAS leverages various types of molecular markers, such as Simple Sequence Repeats (SSRs), Single Nucleotide Polymorphisms (SNPs), and Genotyping-by-Sequencing (GBS), to identify and track quantitative trait loci (QTLs) associated with important agronomic traits (Table 1) (Collard and Mackill, 2008; Song et al., 2023). For instance, the use of GBS has enabled high-throughput genotyping, which is particularly useful for large-scale breeding programs (Collard and Mackill, 2008). This approach not only accelerates the breeding cycle but also enhances the accuracy of selecting traits such as disease resistance, yield, and quality (Miedaner and Korzun, 2012).
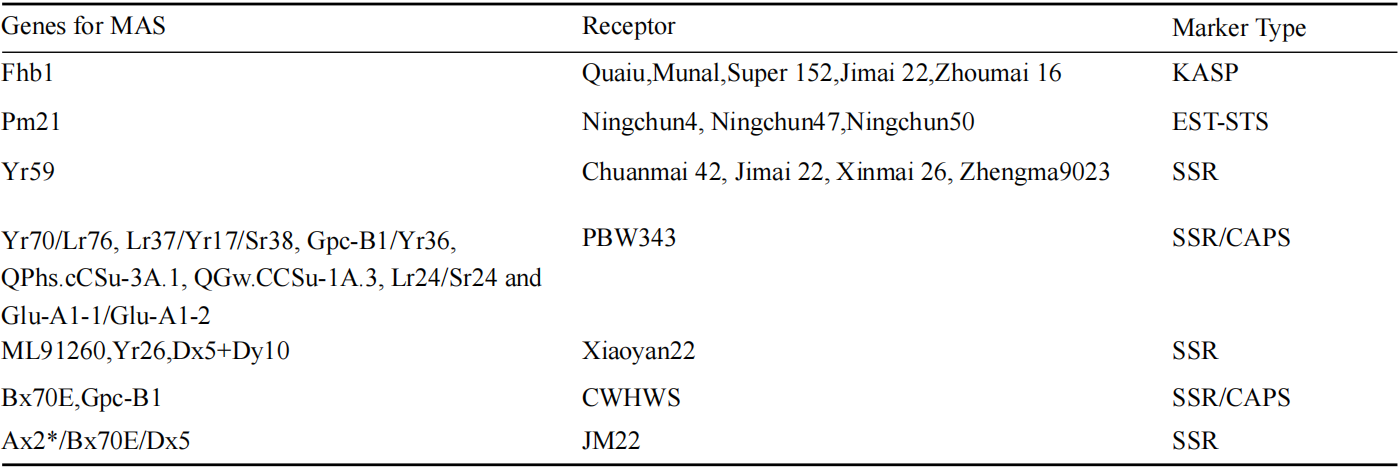 Table 1 Table 1 Examples of using MAS stacking some key genes (Adopted from Song et al., 2023) Table caption: The table presents examples of using marker-assisted selection (MAS) to stack key genes. These examples demonstrate the use of different marker types in various receptor varieties to stack key genes, enhancing breeding efficiency and selection accuracy (Adopted from Song et al., 2023) |
The application of MAS in Triticeae breeding has shown promising results in improving disease resistance. For example, MAS has been successfully used to incorporate resistance genes for wheat rust, barley yellow mosaic viruses, and Fusarium head blight into elite breeding lines (Collard and Mackill, 2008). These advancements highlight the potential of MAS to address some of the most pressing challenges in Triticeae breeding, such as developing varieties that can withstand biotic and abiotic stresses.
4.2 Enhancing crop resilience
Enhancing crop resilience is a critical goal in Triticeae breeding, especially in the face of climate change and increasing biotic pressures. MAS plays a pivotal role in this endeavor by enabling the precise selection of traits that contribute to resilience, such as drought tolerance, disease resistance, and pest resistance (Mohan et al., 1997; Collard and Mackill, 2008). The identification and incorporation of QTLs associated with these traits into breeding programs can significantly improve the resilience of Triticeae crops.
For instance, the use of MAS has facilitated the identification of QTLs for drought tolerance in wheat, allowing breeders to develop varieties that can thrive in water-limited environments (Song et al., 2023). Similarly, MAS has been employed to enhance disease resistance in barley by incorporating QTLs for resistance to powdery mildew and other pathogens (Miedaner and Korzun, 2012). These efforts are crucial for ensuring food security and sustainability in agriculture.
Moreover, the development of high-throughput genotyping platforms, such as SNP arrays and GBS, has further enhanced the ability to select for resilience traits. These technologies allow for the rapid and cost-effective screening of large populations, making it feasible to identify and select for multiple traits simultaneously (He et al., 2014). This holistic approach to breeding can lead to the development of Triticeae varieties that are not only high-yielding but also resilient to a range of environmental stresses.
4.3 Improving yield and quality traits
Improving yield and quality traits is a primary objective in Triticeae breeding, and MAS has proven to be a valuable tool in achieving this goal. The identification and selection of QTLs associated with yield components, such as grain size, number, and weight, have been facilitated by the use of molecular markers (Lande and Thompson, 1990; Hasan et al., 2021). By incorporating these QTLs into breeding programs, breeders can develop varieties with enhanced yield potential.
In addition to yield, quality traits such as protein content, milling quality, and baking quality are also important targets for MAS in Triticeae breeding. For example, the use of MAS has enabled the selection of wheat varieties with improved protein content and gluten strength, which are critical for bread-making quality (Song et al., 2023). Similarly, MAS has been used to enhance malting quality in barley by selecting for QTLs associated with enzyme activity and grain plumpness (Miedaner and Korzun, 2012).
The integration of MAS with traditional breeding methods has also allowed for the simultaneous improvement of multiple traits. This is particularly important in Triticeae breeding, where trade-offs between yield and quality traits often exist. By using MAS to select for favorable alleles at multiple loci, breeders can develop varieties that achieve a balance between high yield and superior quality (Lande and Thompson, 1990; Collard et al., 2008).
In conclusion, the application of MAS in Triticeae breeding has significant implications for improving yield and quality traits. The ability to precisely select for desirable traits at an early stage, combined with the use of high-throughput genotyping technologies, has the potential to accelerate the development of high-performing Triticeae varieties. As the field of molecular genetics continues to advance, the role of MAS in Triticeae breeding is expected to become even more prominent, driving further improvements in crop productivity and quality.
5 Challenges and Limitations
5.1 Complexity of quantitative traits
Quantitative traits are inherently complex due to their polygenic nature and the influence of environmental factors. These traits result from the combined effects of multiple genes, known as quantitative trait loci (QTL), and their interactions with the environment. This complexity poses significant challenges in identifying and mapping the individual genes responsible for these traits. For instance, the phenotypic variation observed in quantitative traits is often due to the segregation of alleles at multiple QTLs, which are sensitive to genetic, sexual, and external environments (Mackay, 2001). The intricate interplay between these factors makes it difficult to pinpoint the exact genetic loci responsible for the traits.
Moreover, the genetic architecture of quantitative traits involves not only the main effects of individual QTLs but also their epistatic interactions, where the effect of one gene is modified by one or several other genes. This adds another layer of complexity to the genetic analysis. For example, Bayesian models have been developed to identify multiple QTLs with complex epistatic patterns, demonstrating the need for sophisticated statistical tools to handle the vast number of potential genetic effects (Yi et al., 2003). These models must account for the number, positions, and genetic effects of QTLs, which is a daunting task given the high dimensionality of the data.
5.2 Accuracy and precision in QTL mapping
The accuracy and precision of QTL mapping are critical for the successful identification of genes associated with quantitative traits. However, several factors can affect these parameters. One major issue is the resolution of the mapping process. High-resolution recombination or linkage disequilibrium mapping is required to narrow down the genomic intervals containing the QTLs and to nominate candidate genes (Mackay, 2001). This process is often hampered by the limited number of recombination events in the mapping populations, which can result in large confidence intervals for the QTL positions.
Additionally, the statistical power to detect QTLs and the precision of parameter estimation can be improved by using multiple trait analysis. This approach takes into account the correlated structure of multiple traits, providing a more comprehensive understanding of the genetic basis of the traits (Jiang et al., 1995). However, this method also requires sophisticated statistical models and computational tools, which may not be readily available or easy to implement.
Another challenge is the potential for false positives and false negatives in QTL mapping. The presence of multiple testing issues and the need for stringent significance thresholds can lead to the exclusion of true QTLs or the inclusion of spurious ones. This necessitates the use of robust statistical methods and validation through independent populations or environments to ensure the reliability of the identified QTLs (Lephuthing et al., 2022).
5.3 Translational challenges
Translating the findings from QTL mapping into practical applications in breeding and genetics is fraught with challenges. One of the primary issues is the difficulty in matching the identified QTLs to specific genetic loci and understanding their functional roles. This requires not only high-resolution mapping but also functional validation through genetic and/or functional complementation and gene expression analyses (Mackay, 2001). The process is time-consuming and resource-intensive, often requiring the integration of various types of data and experimental approaches.
Furthermore, the genetic architecture of quantitative traits can vary significantly across different populations and environments. This variability can complicate the transfer of QTL information from one context to another. For example, QTLs identified in one population may not be relevant or may have different effects in another population due to differences in genetic background and environmental conditions (Jansen et al., 2003). This necessitates the validation and fine-tuning of QTLs in multiple populations and environments to ensure their utility in breeding programs.
Another translational challenge is the integration of QTL information into breeding programs. Marker-assisted selection (MAS) and genomic selection (GS) are powerful tools for incorporating QTL information into breeding decisions. However, the implementation of these techniques requires a thorough understanding of the genetic architecture of the traits, the availability of reliable markers, and the development of appropriate breeding strategies (Shariatipour et al., 2021). The complexity of quantitative traits and the need for high-throughput genotyping and phenotyping platforms can pose significant logistical and financial barriers to the widespread adoption of these technologies.
In summary, while QTL analysis holds great promise for advancing our understanding of the genetic basis of quantitative traits and improving breeding programs, several challenges and limitations must be addressed. These include the complexity of the traits, the accuracy and precision of QTL mapping, and the translational challenges of applying QTL information in practical breeding and genetics. Addressing these issues will require continued advancements in statistical methods, computational tools, and experimental approaches, as well as collaborative efforts across disciplines and institutions.
6 Future Directions in Triticeae QTL Research and Breeding
6.1 Advancements in genomic technologies
The field of quantitative trait loci (QTL) analysis in Triticeae has significantly benefited from advancements in genomic technologies. Traditional methods, such as analysis of variance, have evolved to incorporate high-resolution genetic maps and multiple markers, enhancing the power to study and locate multiple interacting QTLs (Doerge, 2002). The introduction of whole-genome resequencing techniques, such as QTL-seq, has revolutionized the rapid identification of QTLs. This method involves sequencing DNA from two bulked populations with extreme trait values, allowing for the efficient mapping of QTLs in a variety of plant species, including rice and potentially Triticeae (Takagi et al., 2013).
Moreover, Bayesian mapping methods have been developed to handle incomplete inbred line cross data, providing probabilistic measures to estimate the number and location of QTLs on chromosomes (Sillanpää and Arjas, 1998). These advancements not only improve the precision of QTL mapping but also facilitate the identification of candidate genes associated with important agronomic traits. For instance, comparative genomic analysis has identified stable QTLs for grain yield, quality traits, and micronutrient contents in wheat, highlighting the potential for these technologies to enhance breeding programs (Shariatipour et al., 2021).
6.2 Integration of multi-omics data
The integration of multi-omics data, including genomics, transcriptomics, proteomics, and metabolomics, represents a promising direction for QTL research in Triticeae. Expression QTL (eQTL) analysis, which treats transcripts as individual phenotypes, has emerged as a powerful tool to understand the molecular basis of quantitative traits (Kliebenstein, 2009). This approach can elucidate the ecological and evolutionary significance of transcript variation and accelerate the identification of genes underlying complex traits.
Additionally, multi-trait analysis models that consider the correlated structure of multiple traits can improve the statistical power and precision of QTL mapping. These models allow for the testing of hypotheses related to genetic correlations between traits, pleiotropy, and QTL by environment interactions, which are crucial for understanding the genetic architecture of complex traits (Jiang and Zeng, 1995). The integration of multi-omics data can provide a comprehensive view of the genetic and molecular networks that control important agronomic traits, facilitating the development of more resilient and high-yielding Triticeae varieties.
6.3 Climate change adaptation
Climate change poses significant challenges to agriculture, necessitating the development of crop varieties that can withstand abiotic stresses such as drought, heat, and salinity. QTL mapping for stress tolerance traits is critical for breeding Triticeae varieties that can adapt to changing environmental conditions. For example, QTLs associated with salt stress tolerance have been identified in wheat, with specific loci linked to traits such as relative water content, membrane stability, and chlorophyll content (Ilyas et al., 2019). These findings highlight the potential for marker-assisted selection to develop salinity-tolerant wheat varieties.
Furthermore, the use of multiparental cross populations in QTL mapping can enhance the genetic basis and mapping resolution for stress tolerance traits. This approach has been successfully applied in durum wheat to identify QTLs for agronomic traits across different environments, demonstrating the prevalence of environment-specific QTLs with small effects (Milner et al., 2016). By leveraging the genetic diversity and adaptability of Triticeae species, breeders can develop varieties that are better equipped to cope with the impacts of climate change.
In conclusion, the future of QTL research in Triticeae lies in the continued advancement of genomic technologies, the integration of multi-omics data, and the focus on climate change adaptation. These directions will not only enhance our understanding of the genetic basis of complex traits but also drive the development of resilient and high-yielding Triticeae varieties, ensuring food security in the face of global environmental challenges.
7 Concluding Remarks
Quantitative Trait Loci (QTL) analysis in Triticeae has significantly advanced our understanding of the genetic basis of important agronomic traits. Various studies have demonstrated the utility of different QTL mapping approaches, such as QTL-seq, multiparental cross designs, and meta-analysis, in identifying QTLs associated with traits like grain yield, disease resistance, and physiological attributes. For instance, QTL-seq has been effectively used in rice to rapidly identify QTLs for traits such as resistance to fungal diseases and seedling vigor. In durum wheat, multiparental cross designs have provided a broader genetic basis and higher mapping resolution, identifying QTLs for yield and other agronomic traits across different environments. Meta-analyses have further refined these findings, consolidating QTLs into meta-QTLs with reduced confidence intervals, thereby enhancing the reliability and stability of QTLs for traits like grain yield and micronutrient content.
Continued research in QTL analysis is crucial for several reasons. It enables the identification of stable and reliable QTLs that can be used in marker-assisted selection (MAS) to improve crop traits efficiently. The integration of advanced genomic tools and techniques, such as whole-genome resequencing and comparative genomics, can further enhance the precision of QTL mapping and the identification of candidate genes. Third, understanding the genetic basis of complex traits through QTL analysis can provide insights into the molecular mechanisms underlying these traits, facilitating the development of superior genotypes with enhanced yield, disease resistance, and stress tolerance. The application of QTL analysis in breeding programs can lead to the development of crops that are better adapted to changing environmental conditions, thereby ensuring food security.
The future of QTL analysis in Triticeae looks promising, with several emerging trends and technologies poised to revolutionize the field. The use of high-throughput sequencing technologies and advanced bioinformatics tools will likely lead to more precise and comprehensive QTL mapping. Additionally, the integration of QTL analysis with other genomic approaches, such as genome-wide association studies (GWAS) and genomic selection, can enhance the accuracy and efficiency of breeding programs. Collaborative efforts and data sharing among researchers will also play a critical role in accelerating the discovery and application of QTLs in crop improvement. As we move forward, it is essential to focus on translating QTL research into practical breeding strategies that can address the challenges of modern agriculture, such as climate change, resource limitations, and the need for sustainable crop production.
Acknowledgments
The authors extend sincere thanks to two anonymous peer reviewers for their feedback on the manuscript.
Conflict of Interest Disclosure
The authors affirm that this research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Backes G., Graner A., Foroughi-Wehr B., Fischbeck G., Wenzel G., and Jahoor A., 1995, Localization of quantitative trait loci (QTL) for agronomic important characters by the use of a RFLP map in barley (Hordeum vulgare L.), Theoretical and Applied Genetics, 90: 294-302.
https://doi.org/10.1007/BF00222217
PMid:24173906
Börner A., Schumann E., Fürste A., Cöster H., Leithold B., Röder M., and Weber W., 2002, Mapping of quantitative trait loci determining agronomic important characters in hexaploid wheat (Triticum aestivum L.), Theoretical and Applied Genetics, 105: 921-936.
https://doi.org/10.1007/s00122-002-0994-1
PMid:12582918
Collard B., and Mackill D., 2008, Marker-assisted selection: an approach for precision plant breeding in the twenty-first century, Philosophical Transactions of the Royal Society B: Biological Sciences, 363: 557-572.
https://doi.org/10.1098/rstb.2007.2170
PMid:17715053 PMCid:PMC2610170
Crepieux S., Lebreton C., Servin B., and Charmet G., 2004, Quantitative trait loci (QTL) detection in multicross inbred designs, Genetics, 168: 1737-1749.
https://doi.org/10.1534/genetics.104.028993
PMid:15579720 PMCid:PMC1448798
Doerge R., 2002, Multifactorial genetics: Mapping and analysis of quantitative trait loci in experimental populations, Nature Reviews Genetics, 3: 43-52.
https://doi.org/10.1038/nrg703
PMid:11823790
Guan P., Lu L., Jia L., Kabir M., Zhang J., Lan T., Zhao Y., Xin M., Hu Z., Yao Y., Ni Z., Sun Q., and Peng H., 2018, Global QTL analysis identifies genomic regions on chromosomes 4A and 4B harboring stable loci for yield-related traits across different environments in wheat (Triticum aestivum L.), Frontiers in Plant Science, 9: 529.
https://doi.org/10.3389/fpls.2018.00529
PMid:29922302 PMCid:PMC5996883
Hasan N., Choudhary S., Naaz N., Sharma N., and Laskar R., 2021, Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes, Journal of Genetic Engineering & Biotechnology, 19(1): 128.
https://doi.org/10.1186/s43141-021-00231-1
PMid:34448979 PMCid:PMC8397809
He J., Zhao X., Laroche A., Lu Z., Liu H., and Li Z., 2014, Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding, Frontiers in Plant Science, 5: 484.
https://doi.org/10.3389/fpls.2014.00484
PMid:25324846 PMCid:PMC4179701
Hoeschele I., Uimari P., Grignola F., Zhang Q., and Gage K., 1997, Advances in statistical methods to map quantitative trait loci in outbred populations, Genetics, 147(3): 1445-1457.
https://doi.org/10.1093/genetics/147.3.1445
PMid:9383084 PMCid:PMC1208265
Huang X., Cöster H., Ganal M., and Röder M., 2003, Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum aestivum L.), Theoretical and Applied Genetics, 106: 1379-1389.
https://doi.org/10.1007/s00122-002-1179-7
PMid:12750781
Ilyas N., Amjid M., Saleem M., Khan W., Wattoo F., Rana R., Maqsood R., Zahid A., Shah G., Anwar A., Ahmad M., Shaheen M., Riaz H., and Ansari M., 2019, Quantitative trait loci (QTL) mapping for physiological and biochemical attributes in a Pasban90/Frontana recombinant inbred lines (RILs) population of wheat (Triticum aestivum) under salt stress condition, Saudi Journal of Biological Sciences, 27: 341-351.
https://doi.org/10.1016/j.sjbs.2019.10.003
PMid:31889856 PMCid:PMC6933172
Jansen R., 2004, Quantitative trait loci in inbred lines, In Balding D.J., Bishop M., and Cannings C., (eds) Handbook of Statistical Genetics , Wiley-Interscience, State of New Jersey, America, pp.587-622.
https://doi.org/10.1002/9780470061619.ch18
PMid:17317349
Jansen R., Jannink J., and Beavis W., 2003, Mapping quantitative trait loci in plant breeding populations, Crop Science, 43: 829-834.
https://doi.org/10.2135/cropsci2003.8290
Jiang C., and Zeng Z., 1995, Multiple trait analysis of genetic mapping for quantitative trait loci, Genetics, 140(3): 1111-1127.
https://doi.org/10.1093/genetics/140.3.1111
PMid:7672582 PMCid:PMC1206666
Kliebenstein D., 2009, Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs, Annual Review of Plant Biology, 60: 93-114.
https://doi.org/10.1146/annurev.arplant.043008.092114
PMid:19012536
Lande R., and Thompson R., 1990, Efficiency of marker-assisted selection in the improvement of quantitative traits, Genetics, 124(3): 743-756.
https://doi.org/10.1093/genetics/124.3.743
PMid:1968875 PMCid:PMC1203965
Lephuthing M., Khumalo T., Tolmay V., Dube E., and Tsilo T., 2022, Genetic mapping of quantitative trait loci associated with plant height and yield component traits in a wheat (Triticum aestivum L.) doubled haploid population derived from tugela-DN × elands, Agronomy, 12(10): 2283.
https://doi.org/10.3390/agronomy12102283
Mackay T., 2001, The genetic architecture of quantitative traits, Annual review of genetics, 35: 303-339.
https://doi.org/10.1146/annurev.genet.35.102401.090633
PMid:11700286
Miedaner T., and Korzun V., 2012, Marker-assisted selection for disease resistance in wheat and barley breeding, Phytopathology, 102(6): 560-566.
https://doi.org/10.1094/PHYTO-05-11-0157
PMid:22568813
Milner S., Maccaferri M., Huang B., Mantovani P., Massi A., Frascaroli E., Tuberosa R., and Salvi S., 2016, A multiparental cross population for mapping QTL for agronomic traits in durum wheat (Triticum turgidum ssp. durum), Plant Biotechnology Journal, 14(2): 735-748.
https://doi.org/10.1111/pbi.12424
PMid:26132599
Mohan M., Nair S., Bhagwat A., Krishna T., Yano M., Bhatia C., and Sasaki T., 1997, Genome mapping, molecular markers and marker-assisted selection in crop plants, Molecular Breeding, 3: 87-103.
https://doi.org/10.1023/A:1009651919792
Paterson A., Deverna J., Lanini B., and Tanksley S., 1990, Fine mapping of quantitative trait loci using selected overlapping recombinant chromosomes, in an interspecies cross of tomato, Genetics, 124(3): 735-742.
https://doi.org/10.1093/genetics/124.3.735
PMid:1968874 PMCid:PMC1203964
Powder K., 2020, Quantitative trait loci (QTL) mapping, Methods in molecular biology, 2082: 211-229.
https://doi.org/10.1007/978-1-0716-0026-9_15
PMid:31849018
Shariatipour N., Heidari B., Tahmasebi A., and Richards C., 2021, Comparative Genomic analysis of quantitative trait loci associated with micronutrient contents, grain quality, and agronomic traits in wheat (Triticum aestivum L.), Frontiers in Plant Science, 12: 709817.
https://doi.org/10.3389/fpls.2021.709817
PMid:34712248 PMCid:PMC8546302
Sillanpää M., and Arjas E., 1998, Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data, Genetics, 148(3): 1373-1388.
https://doi.org/10.1093/genetics/148.3.1373
PMid:9539450 PMCid:PMC1460044
Singh B., and Singh A., 2015, Mapping of quantitative trait loci, Marker-assisted plant breeding: principles and practices, Springer, New Delhi, pp.185-216.
https://doi.org/10.1007/978-81-322-2316-0_7
Song L., Wang R., Yang X., Zhang A., and Liu D., 2023, Molecular markers and their applications in marker-assisted selection (MAS) in bread wheat (Triticum aestivum L.), Agriculture, 13(3): 642.
https://doi.org/10.3390/agriculture13030642
Takagi H., Abe A., Yoshida K., Kosugi S., Natsume S., Mitsuoka C., Uemura A., Utsushi H., Tamiru M., Takuno S., Innan H., Cano L., Kamoun S., and Terauchi R., 2013, QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations, The Plant Journal : for Cell and Molecular Biology, 74(1): 174-183.
https://doi.org/10.1111/tpj.12105
PMid:23289725
Tshikunde N., Mashilo J., Shimelis H., and Odindo A., 2019, Agronomic and physiological traits, and associated quantitative trait loci (QTL) affecting yield response in wheat (Triticum aestivum L.): a review, Frontiers in Plant Science, 10: 1428.
https://doi.org/10.3389/fpls.2019.01428
PMid:31749826 PMCid:PMC6848381
Yi N., and Xu S., 2001, Bayesian mapping of quantitative trait loci under complicated mating designs, Genetics, 157(4): 1759-1771.
https://doi.org/10.1093/genetics/157.4.1759
PMid:11290729 PMCid:PMC1461584
Yi N., Xu S., and Allison D., 2003, Bayesian model choice and search strategies for mapping interacting quantitative trait Loci, Genetics, 165(2): 867-883.
https://doi.org/10.1093/genetics/165.2.867
PMid:14573494 PMCid:PMC1462771
Zhang L., Liu D., Guo X., Yang W., Sun J., Wang D., and Zhang A., 2010, Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat, Journal of Integrative Plant Biology, 52(11): 996-1007.
https://doi.org/10.1111/j.1744-7909.2010.00967.x
PMid:20977657
.png)



. PDF(0KB)
. FPDF(win)
. FPDF(mac)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Guiping Zhang
. Renxiang Cai
Related articles
. Quantitative trait loci (QTL)
. Marker-assisted selection (MAS)
. Triticeae
. Genetic mapping
. Crop improvement
Tools
. Email to a friend
. Post a comment